Thank you for the wonderful tool and efforts in maintaining this community.
We have been very interested in working with denoised single-cell/single-nucleus transcription vectors. Specifically, we are very interested in obtaining the batch-corrected “count” vectors after denoising. We have been using the scVI tool and found the following:
For our project, we have been using scVI to run the raw counts from a cohort with about 12 separately-sequenced individuals (treated as batches). In addition, we included all the genes (rather than just the highly variable genes) because we want as many genes as possible for downstream analysis. We found that the latent space was very well batch-corrected, but the denoised vectors still showed batch-specific clustering. Is there something we should be doing differently, beyond following the tutorial?
Are there any recommendations beyond using the original scVI model? Specifically, are there any updates or more recent approaches in your tool suite that we should consider?
Hi, what do you hope another tool to do? I can then link to other tools within scVI for this purpose. The normalized expression is by default not batch corrected but only when you provide transform_batch the generated counts will be projected to this batch leading to corrected gene expression
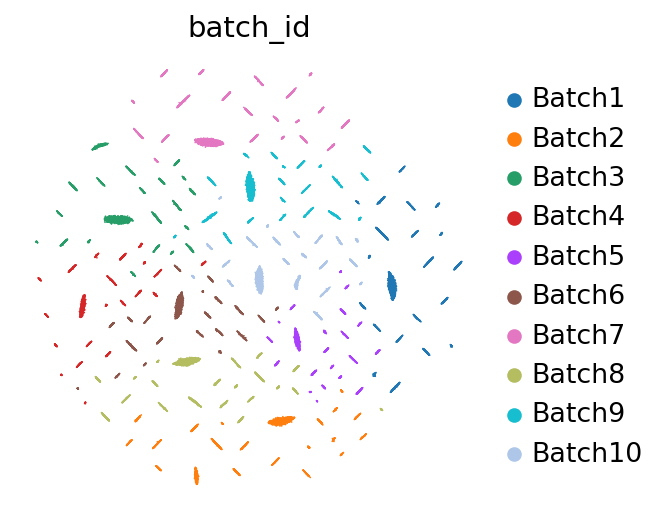
I’m using the Splatter simulated dataset with 10 batches with dropout and aiming to remove batch effects to obtain corrected/denoised read counts. While the scVI model shows a good removal of batch effects in the latent space UMAP, which reflects in meaningful clustering potentially indicative of cell types, the results diverge when examining the denoised read counts obtained via get_normalized_expression().
Here’s the situation:
Using get_normalized_expression(): I’ve experimented with the parameters library_size, n_samples, and transform_batch. When I set transform_batch, only the integer 0 is accepted without errors. Other inputs, like real batch names or other integers, return errors like:
ValueError: “1” not a valid batch category.
ValueError: “Batch1” not a valid batch category.
UMAP Plots: The UMAP plot of denoised counts shows:
A “spaghetti-like” pattern when library_size = "latent"
Both patterns persist regardless of changes to n_samples and transform_batch.
I’m concerned whether I might be misusing the function or if there’s a limitation in scVI concerning the generation of denoised data. I’d appreciate any insights or suggestions on how to proceed from here. Could there be specific adjustments or alternative methods you would recommend for better handling of batch effects and denoised data visualization?
Here are the UMAP plots for reference: Raw Data UMAP:
Maybe you first explain why you are doing that. Splatter contains no gene-gene dependencies (except they have a new simulation) but all genes are sampled independently. This is bad for all embedding methods that try to cover gene-gene dependencies and especially for the generative models. UMAP then strongly manipulates these embeddings and you end up with spaghettis or distinct clusters. Just use real data or more advanced simulations e.g. scDesign?