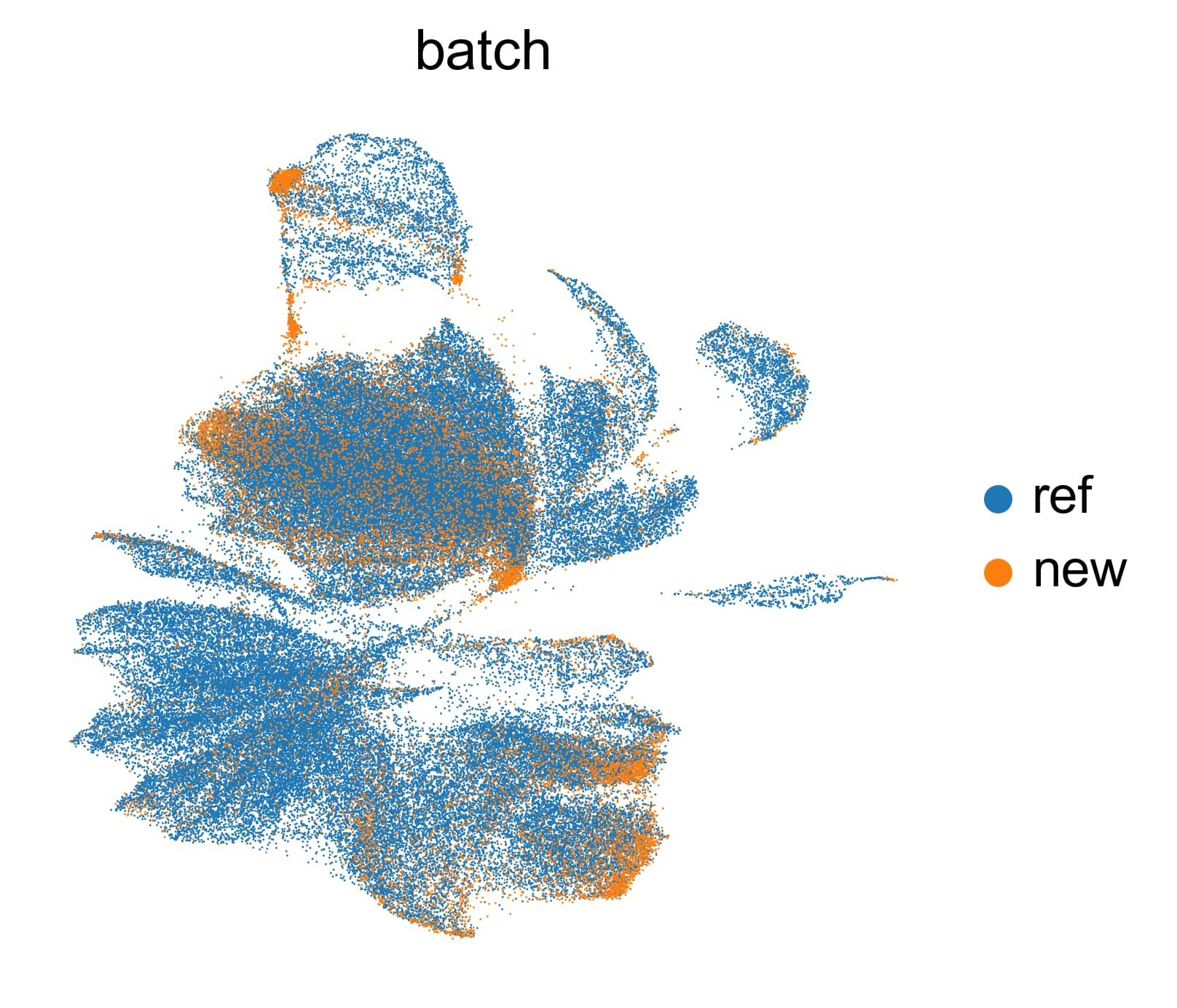
Hello! I’m trying to integrate my query dataset (~13000 cells) into a larger reference dataset (OMG database). I’ve selected cells from E11.5 embryos, matching my sample. I select 5,000 highly variable genes using the pearson residuals. However, my resulting UMAP shows me that they are clearly not integrated at all. I’ve tried downsampling the reference dataset (200,000 cells → 30,000 cells), but it doesn’t help at all. Here’s the UMAP:

Some information about my dataset: it’s generated from a transgenic mouse embryo E11.5 which have been FACSed for mCherry expression. It’s a mix of cell types which I only vaguely know the composition of (should contain primarily brain, heart, limb tissue). Additionally, there are 30 artificial genes which I’ve inserted into the genome (they don’t exist in the WT transcriptome), but they’re promptly removed in the HVG selection.
What I’m wondering is whether this is a problem because SCANVI wasn’t designed for this type of data (my query dataset has a subset of the reference dataset cell types, and potentially in different proportions), or if it’s a problem in how I’ve set up the model/parameters.
Here’s my code:
ref_data = sc.read("/project/omg/adata_JAX_dataset_2_E115.h5ad")
fresh_data = sc.read("/project/fresh_combined-null-1-1_Filtered_ensembl.h5ad")
fresh_data.obs["celltype"] = "Unknown"
# Calculate QC and filter outliers
sc.pp.calculate_qc_metrics(fresh_data, percent_top=None, log1p=False, inplace=True)
fresh_data.obs["outlier_total"] = fresh_data.obs.total_counts > 10000
fresh_data.obs["outlier_ngenes"] = fresh_data.obs.n_genes_by_counts > 1500
sc.pp.filter_genes(fresh_data, min_cells=1)
# Concatenate both datasets
full_data = anndata.concat([ref_data, fresh_data], label="batch", join = "inner")
full_data.obs_names_make_unique()
full_data.obs["batch"] = full_data.obs["batch"].cat.rename_categories(["Ref", "Fresh"])
full_data.layers["counts"] = full_data.X
# Select HVG
sc.experimental.pp.highly_variable_genes(full_data, flavor="pearson_residuals", n_top_genes=5000, batch_key = "batch", subset = True)
# Setup variables
SCVI_LATENT_KEY = "X_scVI"
SCANVI_LATENT_KEY = "X_scANVI"
SCANVI_PREDICTIONS_KEY = "predictions_scanvi"
SCVI_CLUSTERS_KEY = "leiden_scVI"
SCANVI_PREDICTIONS_SCORE = "score_scanvi"
# Train SCVI
sca.models.SCVI.setup_anndata(full_data, layer = "counts", labels_key="celltype")
scvi_full = sca.models.SCVI(
full_data,
n_layers=4,
n_latent=50,
gene_likelihood="nb",
encode_covariates=True,
deeply_inject_covariates=False,
use_layer_norm="both",
use_batch_norm="none",
dropout_rate=0.2,
)
scvi_full.train()
print("Saving SCVI full model.")
scvi_full.save("/project/models/scvi/scvi_omg_115_subsampled30k_integrated_fresh", overwrite=True)
# Train SCANVI
scanvae_full = sca.models.SCANVI.from_scvi_model(scvi_full, unlabeled_category = "Unknown", labels_key = "celltype")
print("Labelled Indices: ", len(scanvae_full._labeled_indices))
print("Unlabelled Indices: ", len(scanvae_full._unlabeled_indices))
scanvae_full.train(max_epochs=10, n_samples_per_label=100)
print("Saving SCANVI full model.")
scanvae_full.save("/project/models/scvi/scanvi_omg_115_subsampled30k_integrated_fresh", overwrite=True)
full_data.obsm[SCANVI_LATENT_KEY] = scanvae_full.get_latent_representation()
full_data.obs[SCANVI_PREDICTIONS_KEY] = scanvae_full.predict()
full_data.obs[SCANVI_PREDICTIONS_SCORE] = scanvae_full.predict(soft = True).max(axis = 1)
full_data.write("/project/omg_115_subsampled30k_integrated_fresh.h5ad", "gzip")
# Get clusters & UMAP
sc.pp.neighbors(full_data, use_rep = SCANVI_LATENT_KEY)
sc.tl.leiden(full_data, key_added=SCVI_CLUSTERS_KEY, flavor = "igraph", n_iterations = 2)
sc.tl.umap(full_data)
sc.pl.umap(full_data, color = ["batch"], frameon = False, ncols = 1)Thanks for any advice!!