Hello ![]() ,
,
I am running scVI VAE tool, with the goal of analyzing both the learned cell embeddings and the reconstructed gene expression. ( i have merfish sample )
I want to verify if the gene ranking is preserved within each reconstructed cell . To do so, i compared the reconstructed profile (R’) vs. Ground Truth (R) for each cell , using Spearman correlation score to assess this rank preservation , and numerous problem arise from that !
the first thing i figure out is that there is a huge bias Toward High Expression/Library Size
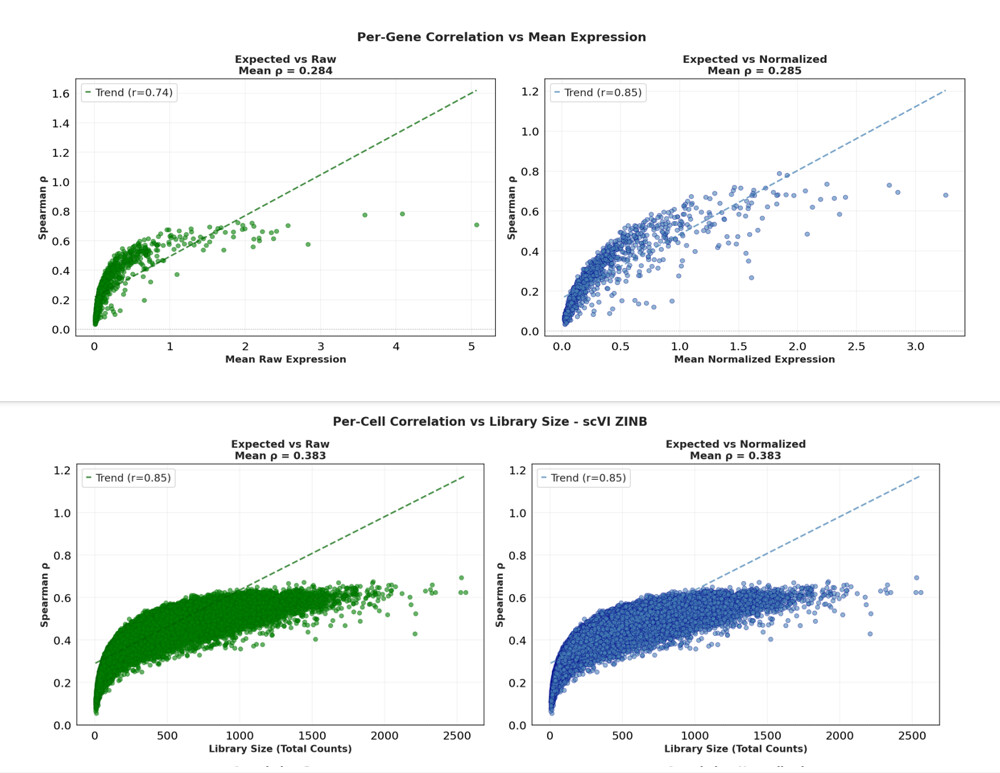
I observed that the model is extremely sensitive to library size and gene abundance: The higher a gene’s expression (and the higher the cell’s library size), the better the ranking is preserved. ( plot 1 & 2 ) . And overall i have a low rank correlation between R’ and R .
It feels like the model focuses only on a subset of high-signal genes (e.g., cell-type markers or pathway specific) to generate embeddings, while ignoring the signal of the majority of other genes.
I attempted to fix this by testing different loss functions (ZINB, NB, Poisson), but unexpectedly, all of them output practically the same spearman correlation metrics, around 0.38
My question are :
1 ) if this Bad ranking for my reconstruction is a normal/expected behavior for scVI/VAEs for all three : zinb/nb/poisson loss ? or Could this problem arise from my specific dataset, or is it likely a hyperparameter/loss function tuning issue?
2 ) Is reconstruction quality is indeed correlated to library size ?
- if i do care about ranking should i discard scvi vae completely ? i think that for the VAE , going for a higher correlation will likely force the model to memorize noise rather than learn biology .
Thank you very much ! i hope my statement was clear , and i am not discarding some crucial insight ![]()
( plot 1 ( per gene correlation ( R’ -R ) based on raw expression )
Plot 2 : per cell correlation based on library size .)